Featured Research

Development of Accurate and Efficient Machine Learning Models
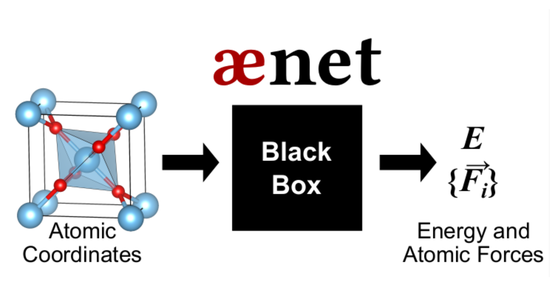
We propose a new method for the efficient training of accurate neural network potentials by including atomic forces via Taylor expansion. The method is demonstrated for water clusters, liquid water, and a solid transition-metal oxide.
A. Cooper, J. Kästner, A. Urban, N. Artrith*, npj Comput Mater 6 (2020) 54 (Open Access).
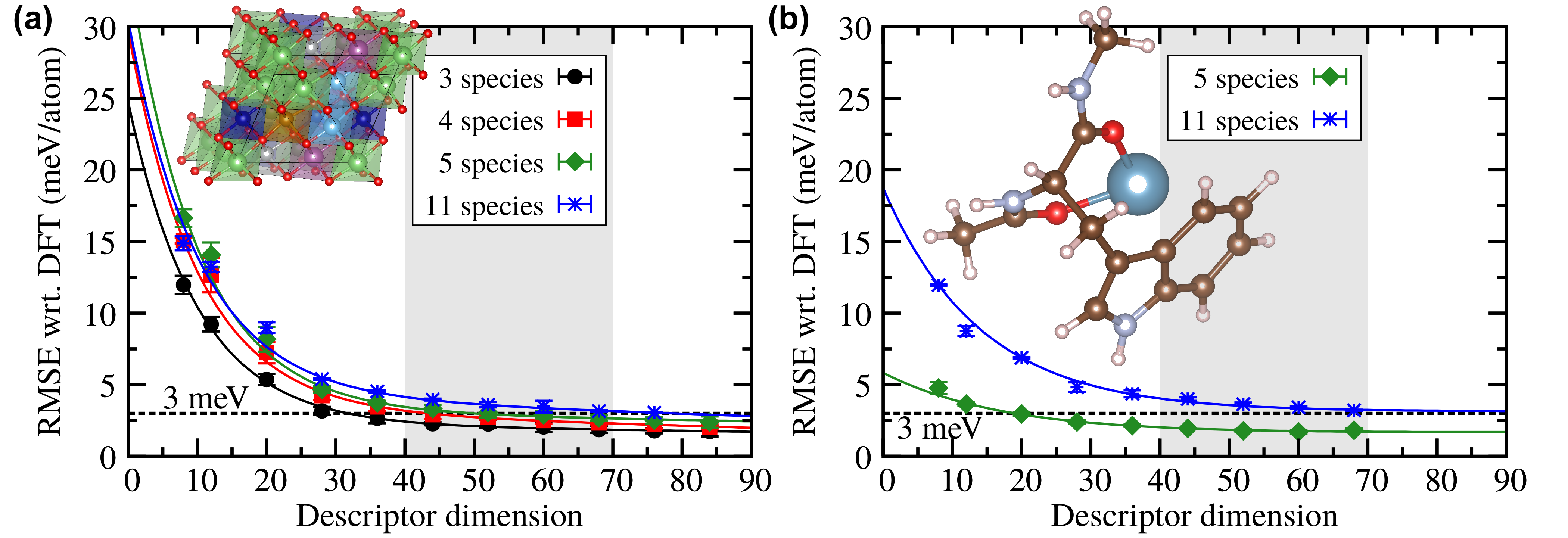
A Novel Descriptor for Machine Learning in Materials Science
We developed a new universal descriptor for general atomic environments based on a simple expansion formalism. This descriptor makes it possible to train atomistic machine learning potentials for materials with many chemical species and is generally useful for structure characterization and data mining.
N. Artrith*, A. Urban, and G. Ceder* Phys. Rev. B 96, 014112 (2017) .
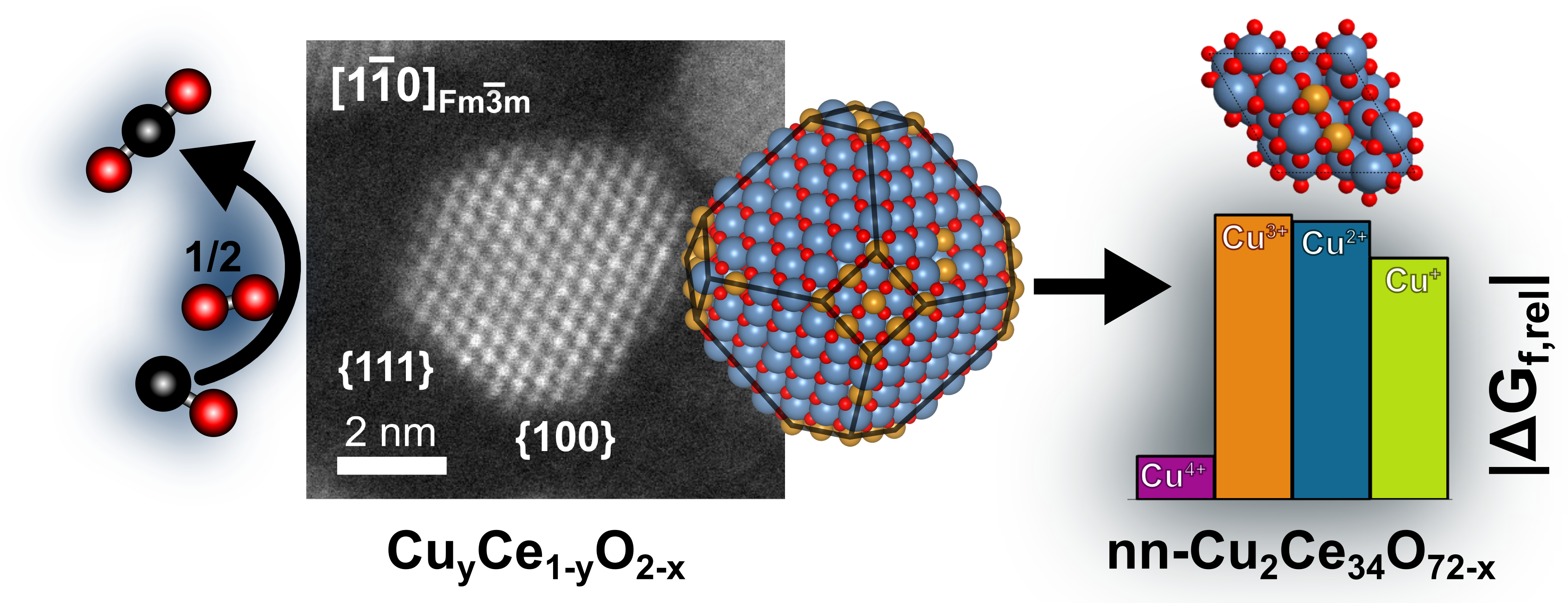
Combining Experiments and Computation to Understand Nanostructured Catalysts
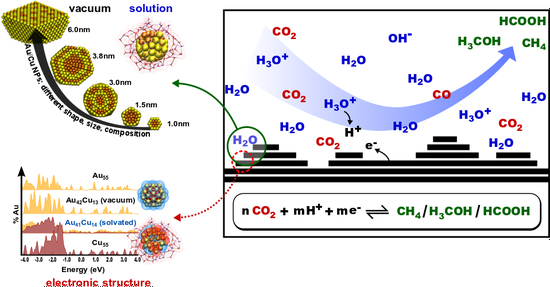
In collaboration with Prof. Yang Shao-Horn’s group, we investigated the nature of the active phase in copper/ceria catalysts for CO oxidation. ANN potential Monte Carlo simulations suggest that Cu(3+) and Cu(2+) preferentially segregate to the {100} surface of the CuyCe1–yO2–x nanoparticle, which is supported by aberration-corrected electron microscopy measurements.
J.S. Elias, N. Artrith, M. Bugnet, L. Giordano, G.A. Botton, A.M. Kolpak, and Y. Shao-Horn*, ACS Catal. 6, 1675-1679 (2016) .
See here a full list of publications .