(Electro-) Catalysis
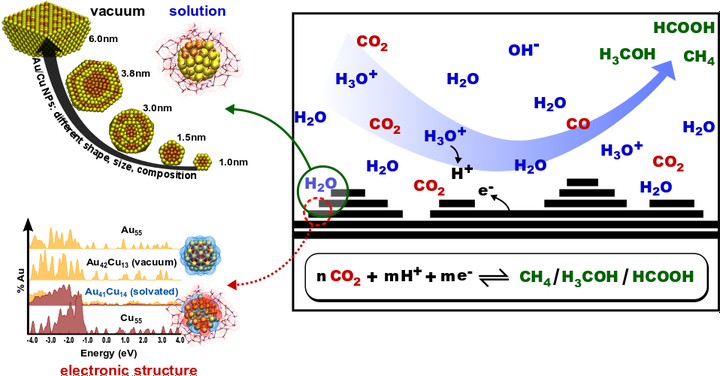 Figure 1. CO2 Conversion over CuAu Nanoparticles [1,2].
Figure 1. CO2 Conversion over CuAu Nanoparticles [1,2].
Heterogeneous catalysis is the foundation of many critical industrial processes, such ammonia or methanol synthesis, and enables (as electrocatalysis) the conversion of clean electric energy to synthetic fuels.
A strong focus of my research has been to elucidate the atomic structure of catalysts, the nature of the catalytically active sites, and the reaction mechanisms using density-functional theory (DFT) and machine learning potentials (MLPs).
Catalytic CuAu Nanoalloys in Solution
To understand the catalytic activity of alloy nanoparticles at the atomic scale, an accurate and efficient method for predicting the thermodynamically stable compositions, shapes and sizes of catalyst particles under varying conditions is needed (see figure). Then the fundamental properties can be determined that make the system active by examining catalytic behavior on the realistic (non-idealized) catalyst surfaces identified in this way.
We employed a combined approach of atomistic reactive MLPs and DFT. MLPs allow the simulation of large realistic structures containing thousands of atoms and enable the extensive sampling of the configuration space. Subsequently, the thermodynamic phase diagram of the catalyst structure and stoichiometry can be determined as a function of temperature and chemical potential. Once important structures and possible reactive sites have been identified, DFT calculations of periodic model structures with a smaller number of atoms in the supercell provide the necessary insight into the electronic structure, thereby providing a set of guidelines for predicting and designing novel earth-abundant replacements with comparable or enhanced activity.
[1] N. Artrith*, and A.M. Kolpak,
Nano Lett., 5, 2670
(2014)
.
[2] N. Artrith*, and A.M. Kolpak,
Comput. Mater. Sci., 110, 20 (2015)
.
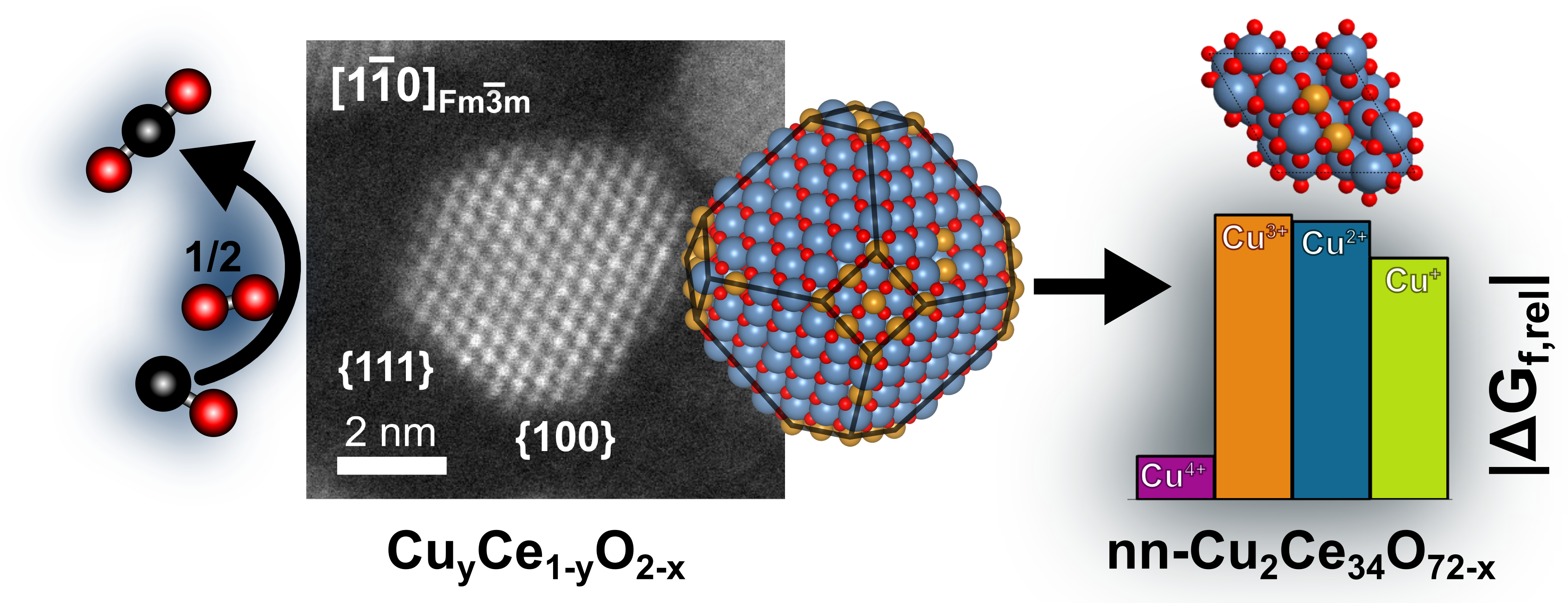
Other Examples from Computational Catalysis
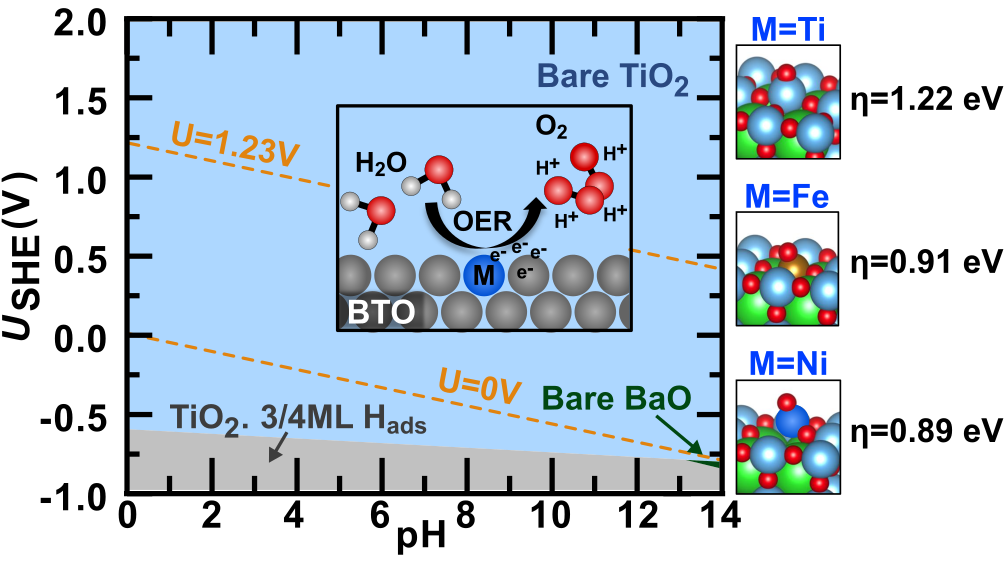
We predicted the atomic ordering in doped ceria nanoparticles (catalysts for CO2 conversion) via MLP-based Monte-Carlo simulations (Fig. 2) [3], and we determined the surface composition of modified BaTiO3 electrocatalyst particles at reaction conditions for water splitting using surface Pourbaix diagrams based on the Nernst equation and DFT computations (Fig. 3) [4].
Another example is the simulation of non-ideal equilibrated Cu@ZnO interface structures for methanol synthesis (Fig. 4). The reaction free energies over catalyst surfaces at room temperature can often be well approximated by entropy-corrected DFT energies of the reaction intermediates.

Select publications related to catalysis
[3] J.S. Elias, N. Artrith, M. Bugnet, L. Giordano, G.A. Botton, A.M. Kolpak, Y. Shao-Horn∗,
ACS Catalysis, 6, 1675 (2016)
.
[4] N. Artrith∗, W. Sailuam, S. Limpijumnong, and A.M. Kolpak,
Phys. Chem. Chem. Phys., 18, 29561 (2016)
.
[5] N. Artrith, B. Hiller, and J. Behler∗,
Phys. Status Solidi B, 250, 1191 (2013)
.
[6] S. Wannakao, N. Artrith, J. Limtrakul, A.M. Kolpak∗,
J. Phys. Chem. C, 121, 20306 (2017)
.
[7] S. Wannakao, N. Artrith, J. Limtrakul, A.M. Kolpak∗,
ChemSusChem, 8, 2745 (2015)
.